Introduction
1 Goals
Structural Bioinformatics (SB) is a broad discipline that encompasses data resources, algorithms, and tools for investigating, analyzing, predicting, and interpreting biomacromolecular structures (Paiva et al. 2022). In this course, we will specifically focus on protein structural bioinformatics, including visualization and analysis of the structure of biomacromolecules as well as the prediction of protein structures and complexes. The premise of SB is that high-resolution structural information about biological systems enables precise reasoning about their functions and the effects of modifications and perturbations.
The goals of SB require at least four different research lines (see Chapter 1 in Gu and Bourne (2009)):
Visualization: Dealing with one or many complex structures and integrating various sources of information such as sequences, structural data, electrostatic fields, locations of functional sites, and areas of variability.
Classification: Grouping similar structures hierarchically to identify common origins and diversification paths. Similar to other fields of biology classification is tedious but required to understand the structural space.
Prediction of structures remains an area of keen interest and a field of research itself. As we will see below, the number of different sequences is much higher than the availability of structures, which make prediction an essential and useful tool.
Simulation. Experimentally obtained structures are primarily static structural models (see warning below). However, the properties of these molecules are often the results of their dynamic motions. The definition of energy functions that govern the folding of proteins and their subsequent stable dynamics can be analyzed by molecular dynamics simulations, although computation capacities may be limiting to reach a biologically relevant timescales.
Powered by vast amounts of data and significant technical advances, the field has undergone a substantial transformation over the past twenty years The enhancement of experimental capabilities to analyze the structure of proteins and other biological molecules and structures (see Callaway (2020)) and the advancement of Artificial Intelligence (AI)-assisted structure prediction have substantially increased the ability of life-science researchers to address various questions concerning protein diversity, evolution, and function. This transformation has boosted in the last 5 years, and its implications for biology, biotechnology, and biomedicine remain largely unpredictable.
2 Before going forward: Protein Structure 101
While it is possible to perform some protein modeling without expertise in structural biology, having a basic understanding of protein structure is highly recommended. In this course, there are often students who do not have a strong background in biology. Additionally, it has been observed that graduate students in biology, biomedicine, and related fields possess varying levels of knowledge about protein structure. For those looking to review or update their understanding of protein structure, it is suggested to read Chapter 2 of Gu and Bourne (2009), the great review by Stollar and Smith (2020) and the Wikipedia and Proteopedia articles on protein structures, which were the primary sources for this brief section (follow picture links).

Proteins are essential components of life, involved in various vital functions as structural elements, scaffolding elements, or active enzymes that catalyze metabolic reactions. Proteins are composed of polymers of amino acids, and the sequence of amino acids of a particular protein is referred to as the Primary structure of the protein. Amino acid chains can spontaneously fold into three-dimensional structures, mainly stabilized by hydrogen bonds between amino acids. The amino acid sequence determines different layers of 3D structure. Each of the 20 natural amino acids has specific physicochemical properties that influence its preferred conformation. Therefore, the initial level of folding is known as the Secondary structure, which forms common patterns as will be discussed further. These segments of secondary structure patterns are capable of folding into three-dimensional forms due to interactions between the side chains of amino acids, known as protein Tertiary structure. Additionally, two or more individual peptide chains can aggregate to form multisubunit proteins, referred to as having Quaternary structure.

It is important to note that the peptide bond itself does not permit rotation as it possesses partial double bond characteristics. Hence, rotation is restricted to the bonds between the Cα and the C = O group (the phi (φ) angle) and the Cα and the NH group (the psi (ψ) angle). The polypeptide backbone chain thus consists of a repeating sequence of two rotatable bonds followed by one non-rotatable (peptide) bond. However, not all 360º of the φ and ψ angles are feasible due to potential steric clashes between neighboring sidechains. For specific angles and amino acid combinations, spatial constraints prevent atoms from occupying the same physical location, which partially accounts for the varying propensities of certain amino acids to adopt different types of secondary structures.

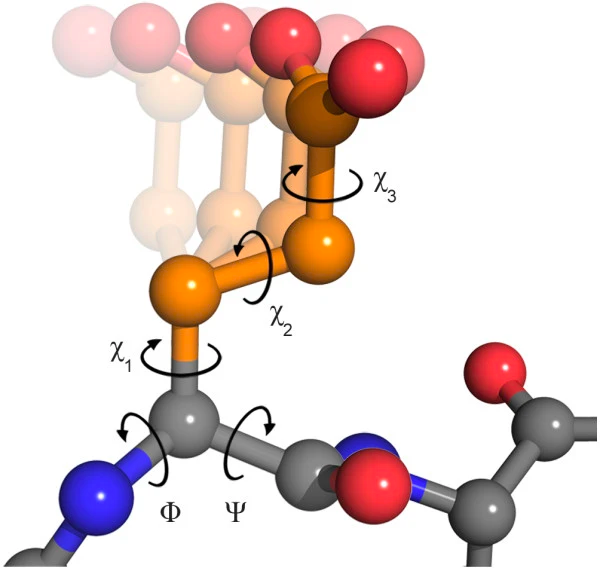
Furthermore, the side chains of amino acids possess their own torsion angles, known as χ1, χ2, χ3, and so forth (Figure 6). These torsion angles significantly influence the secondary and, particularly, the tertiary structures of proteins. The various combinations of side-chain torsions defined by χ angles are referred to as rotamers.
Within these constraints, the two primary local conformations that avoid steric hindrance and maximize backbone-backbone hydrogen bonding are the α-helix and the β-sheet secondary structures. Linus Pauling initially proposed the α-helix as left-handed in 1951, but the myoglobin crystal structure in 1958 revealed that the right-handed form is more common. In the typical right-handed helices, the backbone NH group hydrogen bonds to the backbone C=O group of the amino acid located four residues earlier along the protein sequence. This regular coil shape has the R-groups pointing outward, away from the peptide backbone, and requires about 3.6 residues to complete a full turn of the helix (Figure 4).


Different amino-acid sequences have varying tendencies to form α-helical structures. Methionine, alanine, leucine, glutamate, and lysine have especially high helix-forming propensities, whereas proline and glycine have poor helix-forming propensities. Proline often disrupts or kinks a helix because it lacks an amide hydrogen to form hydrogen bonds and its bulky side chain interferes with the preceding turn’s backbone. Glycine with only a hydrogen as its R-group, is too flexible and entropically costly to maintain the constrained α-helical structure, making it an α-helix breaker.
β-Sheets (Figure 5) consist of two or more extended polypeptide chains called β-strands running alongside each other in either a parallel or antiparallel arrangement. In a β-sheet, residues arrange themselves in a zigzag pattern with adjacent peptide bonds pointing in opposite directions. The NH group and the C=O group of each amino acid form hydrogen bonds with the C=O group and NH group, respectively, on adjacent strands. The chains can run in opposite directions (antiparallel β-sheet) or the same direction (parallel β-sheet). Sidechains from each residue alternate in opposite directions, giving β-sheets hydrophilic and hydrophobic faces, often forming a pattern of alternating hydrophilic and hydrophobic residues in the primary structure.
Large aromatic residues (tyrosine, phenylalanine, tryptophan) and β-branched amino acids (threonine, valine, isoleucine) are commonly found in β-strands. As in the case of α-helixes, β-strands are often ended by glycines, which are especially common in β-turns (the most common connector between strands), as amino acids with positive φ angles.
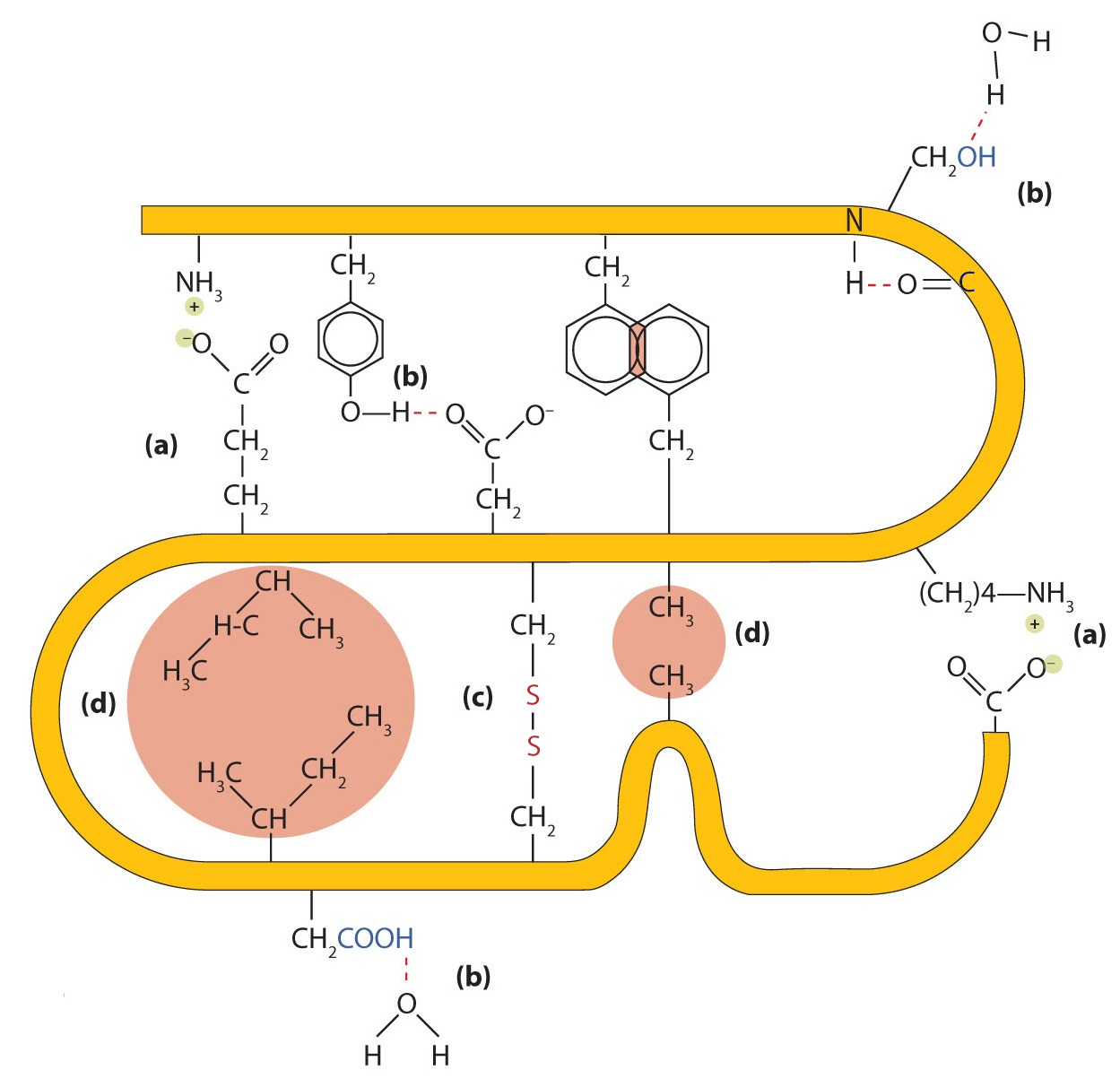
Protein secondary, tertiary, and quaternary structures are maintained by interactions between amino acids (Figure 7). These interactions are typically classified into four types:
- Ionic bonding: Ionic bonds arise from electrostatic attractions between positively and negatively charged amino acid side chains. For example, the attraction between an aspartic acid carboxylate ion and a lysine ammonium ion helps stabilize a specific folded region of a protein.
- Hydrogen bonding: Hydrogen bonds form between a highly electronegative oxygen or nitrogen atom and a hydrogen atom attached to another oxygen or nitrogen atom, such as those in polar amino acid side chains. Hydrogen bonds are crucial for both intra- and intermolecular interactions in proteins, like in alpha-helixes.
- Disulfide linkages. When two cysteine amino acids come close together during protein folding under appropriate redox conditions, oxidation can link their sulfur atoms, forming a disulfide bond. Unlike ionic or hydrogen bonds, these are covalent links, and are thus a classic example of a spontaneus reaction, occurring as a post-translational modification. Although they are sensitive to reducing agents, greatly stabilize the tertiary structure and are vital for the quaternary structure of many proteins, like antibodies.
- Hydrophobic interactions. Dispersion forces arise when a normally nonpolar atom becomes momentarily polar due to an uneven distribution of electrons, leading to an instantaneous dipole that induces a shift of electrons in a neighboring nonpolar atom. Dispersion forces are weak but can be important when other types of interactions are either missing or minimal. The term hydrophobic interaction is often misused as a synonym for dispersion forces. Hydrophobic interactions arise because water molecules engage in hydrogen bonding with other water molecules (or groups in proteins capable of hydrogen bonding). Because nonpolar groups cannot engage in hydrogen bonding, the protein folds in such a way that these groups are buried in the interior part of the protein structure, minimizing their contact with water.
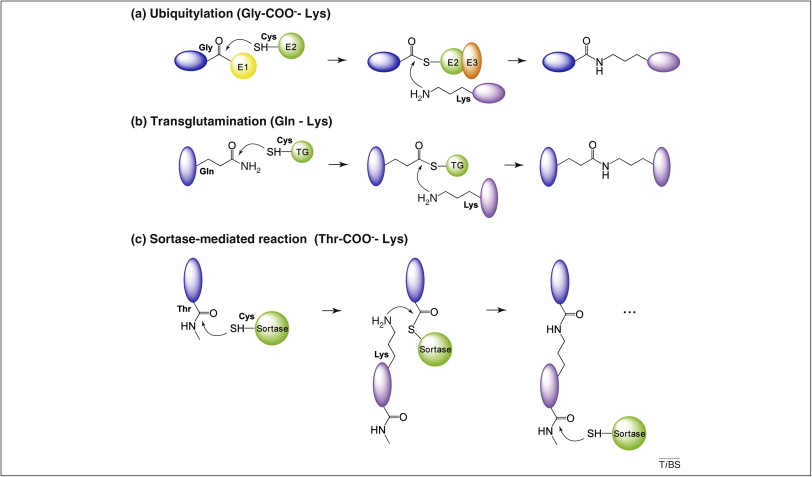
Other less frequent intramolecular interactions might be relevant in some proteins, like the so-called Isopeptide bonds, formed between two protein groups, at least one of which is not an α-amino or α-carboxy group. Examples include ubiquitylation, sumoylation, transglutamination, sortase-mediated cell surface protein anchoring and pilus formation (Kang and Baker 2011). As detailed in Figure 8, all these processes share several feature:
- All involve the reaction of a lysine ε-amino group on one protein with the main α-carboxy group on another protein, except for transglutamination, where the lysine targets a glutamine side chain carboxyamide group.
- All processes are enzyme-mediated and involve a transient thioester intermediate formed by the active site’s cysteine. This intermediate is resolved through nucleophilic attack by the lysine ε-amino group, which completes the bond formation.
In contrast to these enzyme-dependent processes, cross-linking isopeptide bonds form autocatalytically in S. pyogenes major pilin Spy0128 and other Gram+ cell surface proteins, as well as in HK97 phage capsid. In this case, the bond-forming reaction is a proximity-induced reaction that happens when participating amino acids are positioned together in a hydrophobic environment, either through protein folding concurrent with peptide bond formation at the ribosome or by capsid rearrangement (in HK97).
Manipulating inter- and intramolecular amino acid interactions is common for altering protein properties. A classic example is adding nearby cysteines to form disulfide bonds, enhancing stability. Recently, Spy0128 motifs and unnatural reactive amino acids have enabled applications like creating protein staples, thermostable antibodies, and surfaces/nanoparticles that capture proteins (Cao and Wang 2022).
2.1 Ramachandran Plot
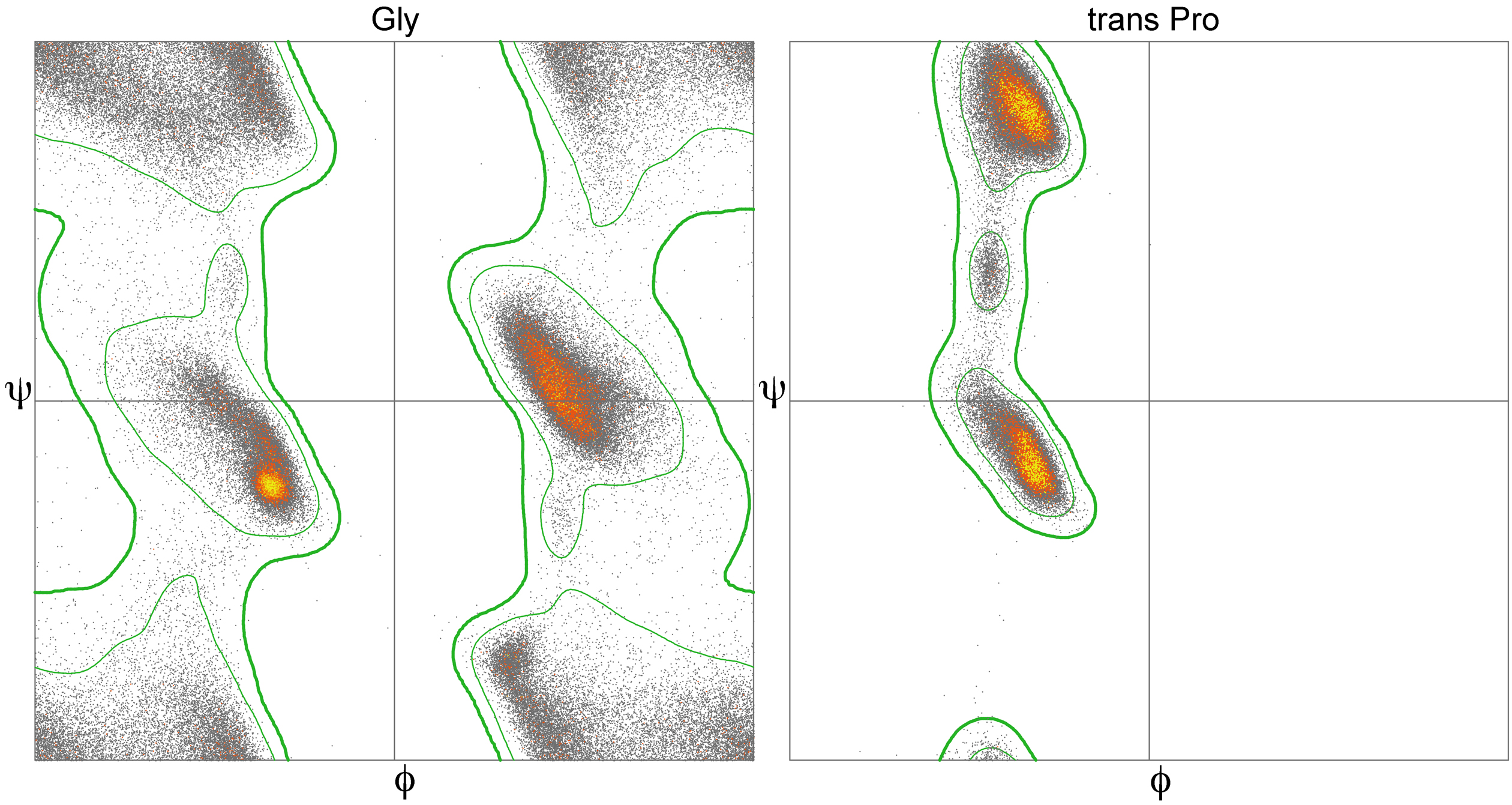
As you likely deduced already, many combinations of φ and ψ angles are prohibited due to the principle of steric exclusion, which dictates that two atoms cannot occupy the same space simultaneously. This concept was initially demonstrated by Gopalasamudram Ramachandran, who developed a plot to visualize the permissible angle values, known as the Ramachandran plot. This plot can display the angles of a specific amino acid, all amino acids in a single protein, or even across many proteins. Analysis of φ and ψ angles in known proteins reveals that approximately three-quarters of all possible φ, ψ combinations are not allowed (Figure 9) that correspond with common secondary structure motifs (Figure 10).


Functionally relevant residues are more likely than others to have torsion angles that plot to the allowed but disfavored regions of a Ramachandran plot. The specific geometry of these functionally relevant residues, while somewhat energetically unfavorable, may be important for the protein’s function, catalytic or otherwise. Such conformations need to be stabilized by the protein using H-bonds, steric packing, or other means, and should very seldom occur for highly solvent-exposed residues.
2.2 Contact maps
A protein contact map illustrates the interactions between all possible pairs of amino acid residues in a protein’s three-dimensional structure. This is displayed as a binary matrix with n rows and columns, where n represents the number of residues in the sequence. In this matrix, the element at position ij is marked as 1 if residues i and j are in contact within the structure. Contact is typically defined as residues being closer than a certain distance threshold, which is 9 Å in the examples shown in Figure 12. The patterns in these maps highlight the differences between motifs and reflect the stretches of secondary structure.

2.3 Protein folds, domains and motifs
The global three dimensional tertiary structure of a protein is commonly referred as its fold. However, assignment of fold is not trivial since there is no single definition of a fold. The term fold defines three major aspects of protein structure, the secondary structure elements of which it is composed, their spatial arrangement and their connectivity. Thus, the term common fold is used to describe the consensus subset of structural elements shared by a group of proteins Andreeva (2012).
Within the overall protein fold, we can recognize distinct domains and motifs.
Domains are compact sections of the protein that represent structurally and (usually) functionally independent regions. Generally, the concept of protein domain can be established from different criteria:
Structural domain: A compact region of structure that is semi-independent of the rest of the protein. This region usually contains and hydrophobic core and can consist of one or more segments of the polypeptide chain, the entire polypeptide chain or several polypeptide chains.
Evolutionary domain: A region of protein that occurs in nature either in isolation or in more than one context of multidomain proteins.
Functional domain: A region of protein structure that is associated with a particular function.
Remarkably, it should be noted that not all these criteria can be simultaneously satisfied. Many conserved structural domains, for instance, may not be associated with a known function and are classified as DUF (domain of unknown function). Likewise, some functional domains can contain more than one structural domains Figure 13
For protein structure analysis and structure comparison purposes, in this course we will mainly refer to the structural definition of protein domains.
On the other hand, motifs are small regions within a protein, which can be defined as sequence or structure. The conservation of amino acids in one small stretch can be strict, as in the case of catalytic residues, or may be defined within a certain group of residues, e.g., hydrophobic, polar, small, charged, etc. Structural motif can be regarded as a few secondary structural elements with a specific geometric arrangement. Indeed, motifs can be also referred as super-secondary structure.
Similar sequence and structural motifs can be found in structurally distinct proteins. This can be of great interest, as they are frequently linked to a function. For example, the nucleophile elbow and oxianion hole is an structural motif that encompasses a discontinuous β/βα motif and constitute the catalytic site in diverse enzymes with esterase activity. See the structures bellow and compare them to find this motif, highligthed in Figure 14.
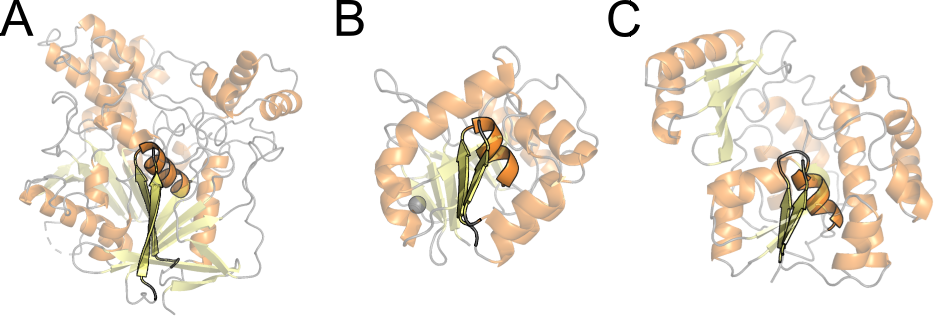
2ack), (B) malonyl-CoA:acyl carrier protein transacylase (pdb 1mla), (C) aspartyl dipeptidase (pdb 1fye).
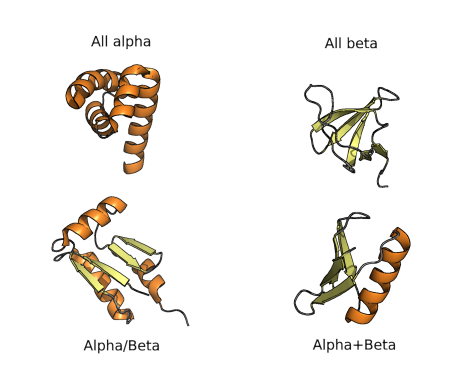
The diversity of protein folds, domains and motifs, and combination of those, can be used for classification of protein structures hierarchically, as in many other fields of biology. The first classification was proposed in the 70’s and consisted of four groups of folds, as shown in the figure below. All α proteins are based almost entirely on an α-helical structure, and all β-structure are based on β-sheets. α/β structure is based on as mixture of α-helices and β-sheet, often organized as parallel β-strands connected by α-helices. On the other hand, α+β structures consist of discrete α-helix and β-sheet motifs that are not interwoven (as they are in α/β proteins). Finally, small proteins span polypeptides with no or little secondary structures.
2.4 Quick exercises
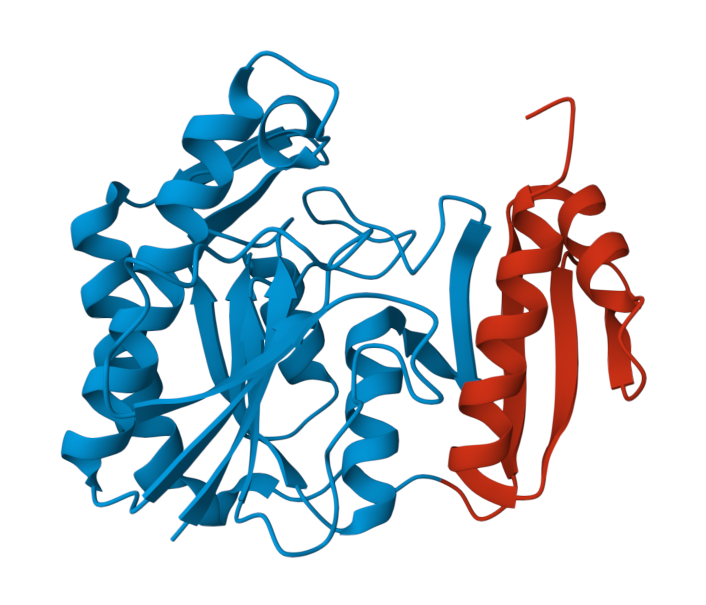
1. Explore the structure below and try to understand its structure. Then answer the question.
How many unique protein chains are there is this protein?As you can see, as the protein is larger, classification gets more difficult. Moreover, as fold space has become more and more complex, these types of classifications have been adjusted and extended such that a complete hierarchy is created. The most commonly referred approaches to this sort of classification are those used by SCOP and CATH databases, as we will see in the Structural Databases section.
2. Playing with secondary structures

There are a few online alternatives to model any peptide sequence and quickly see the effect of amino acid composition in the secondary structure. One of the best-known is Foldit (www.fold.it, Miller et al. (2020)), a gaming platform for biochemistry and structural biology teaching. It is a highly recommended alternative for most courses related to protein structure.
In this course we are going to try a more recent proposal, twitted by Sergey Ovchinnikov:

This is based on ColabFold (see https://github.com/sokrypton/ColabFold and Mirdita et al. (2022)), an Alphafold2 (see Jumper et al. (2021)) free notebook in Google Colab notebook. All you need is a Google account and the cheatsheet in Figure 16.
Now go to ColabFold Single: https://colab.research.google.com/github/sokrypton/af_backprop/blob/beta/examples/AlphaFold_single.ipynb
Construct some small proteins and compare the output. Use 6 recycle steps to obtain diverse structures and then click in the “Animate trajectories” chunk. Note that the first model will take 2-4 min, but the others will be faster. I provide here some interesting examples (IUPAC one-letter amino acid code):
AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
KKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKKK
PVAVEARENGRLAVRVEGAIAVLIRENGRLVVRVEGG
PELEKHREELGEFLKKETGIAVEIRENGRLEVRVEGYTDVKIEGGTERLKRFLEEL
ACTWEGNKLTCA
1. Answer the following questions:
- Why is a poly-K more stable (dark blue) than a poly-A?
- Could you predict the structure of a poly-V or a poly-G?
- What would happen if you introduce a K5W in the structure number 2? and in the 4?
2. Now, try to create peptides with a custom motif, such as:
- Two helices.
- A four-strands beta-sheet.
- Alpha-beta-beta-alpha.