State-of-the-art protein modeling (in 2022)

The recent history of protein structure modeling telling by a contest (CASP)
Every two years since 1994, structural bioinformatics groups carry out a worldwide experiment, predicting a set of unknown protein structures in a controlled, blind-test-like competition and comparing their output with the experimentally obtained structure. This is the CASP or Critical assessment of Protein Structure Prediction.

The best research groups in the field test their new methods and protocols in CASP. However, in CASP13 (2018) an AI company called Deepmind (Google Subsidiary) entered in the scene. Their method, named Alphafold (Senior et al. 2020) clearly won CASP13. Alphafold implemented some improvements in a few recently used approaches, creating a new whole pipeline. Basically, instead of create contact maps from the alignment to then fold the structure, they used a MRF unit (Markov Random Field) to extract in advance the main features of sequence and the MSA and process all of that info into a multilayer NN (called ResNet) that provides the distant map and other information. Then, Alphafold uses all the possibly obtained information to create the structure and then improve it by energy minimization and substitution of portions with a selected DB of protein fragments.

After Alphafold, similar methods were also developed and made available to the general public, like the trRosetta from Baker lab (Yang et al. 2020), available in the Robetta server. This led to some controversy (mostly on Twitter) about the open access to the CASP software and later on DeepMind publishes all the code on GitHub.
CASP14 or when protein structure prediction come to age for (non structural) biologists
In CASP14 the expectation was very high and the guys from DeepMind did not disappoint anyone. Alphafold2 highly outperformed all competitors, both in relative (score respect the other groups) and absolute terms (lowest alpha-C RMSD). As has been highlighted, the accuracy of many of the predicted structures was within the error margin of experimental determination methods (see for instance Mirdita et al. 2022).


Alphafold took some time (eight months, an eternity nowadays) to publish the method (Jumper et al. (2021)) and making it available on Github, but other new methods, like RoseTTAfold (Baek et al. (2021)) and C-I-Tasser (Zheng et al. (2021)) could reproduce their results and were available on public servers, which may have push Deepmind to make everything available to the scientific community (see the Grace Huckins’ article on Wired). Not surprisingly (at least for me), a group of independent scientists (Sergey Ovchinnikov, Milot Mirdita, and Martin Steinegger), decided to implement Alphafold2 in a Colab notebook, named ColabFold Mirdita et al. (2022), freely available online. Other free implementations of Alphafold have been and are available, but ColabFold has been the most widely discussed and known. They implemented some tricks to accelerate the modeling, mainly the use of MMSeqs2 (developed by Martin Steinegger’s group) to search for homolog structures on Uniref30, which made Colabfold a quick method that made all the previous advanced methods almost useless. This was the real breakthrough in the protein structure field, making Alphafold2 available to every one and, also very important, facilitate the evolution of the method, implementing new features, like the prediction of protein complexes ( Evans et al. (2022)), which was actually mentioned first on Twitter.
Alphafold2 as the paradigm of a New Era
Why is Alphafold2 so freaking accurate?
The philosophy behind Alphafold and related methods is treating the protein folding problem as a machine learning problem, kind of of image processing. In all these problems, the input to the Deep Learning model is a volume (3D tensor). In the case of computer vision, 2D images expand as a volume because of the RGB or HSV channels. Similarly, in the case of distance prediction, predicted 1D and 2D features are transformed and packed into 3D volume with many channels of inter-residue information (Pakhrin et al. 2021).
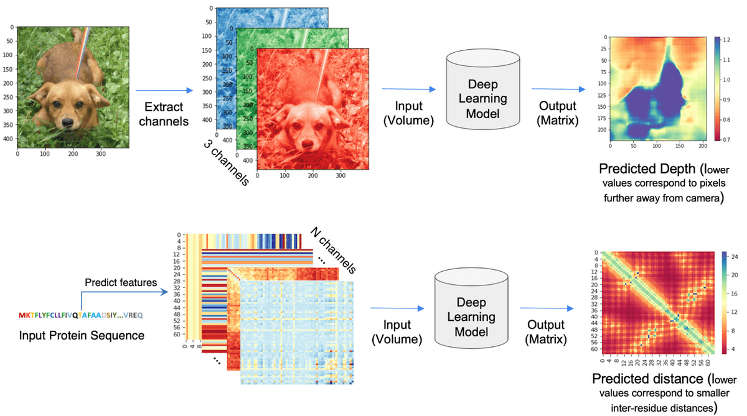
Alphafold2 can be explained as a pipeline with three interconected tasks (see picture below). First, it queries several databases of protein sequences and constructs an MSA that is used to select templates. In the second part of the diagram, AlphaFold 2 takes the multiple sequence alignment and the templates, and processes them in a transformer. This process has been referred by some authors as inter-residue interaction map-threading (Bhattacharya et al. 2021). The objective of this part is to extract layers of information to generate residue interaction maps. A better model of the MSA will improve the network’s characterization of the geometry, which simultaneously will help refine the model of the MSA. Importantly, in the AF2 Evoformer, this process is iterative and the information goes back and forth throughout the network. At every recycling step, the complexity of the map increases and thus, the model improves (the original model uses 3 cycles). As explained in the great post from Carlos Outerial at the OPIG site:
This is easier to understand as an example. Suppose that you look at the multiple sequence alignment and notice a correlation between a pair of amino acids. Let’s call them A and B. You hypothesize that A and B are close, and translate this assumption into your model of the structure. Subsequently, you examine said model and observe that, since A and B are close, there is a good chance that C and D should be close. This leads to another hypothesis, based on the structure, which can be confirmed by searching for correlations between C and D in the MSA. By repeating this several times, you can build a pretty good understanding of the structure.
The third part of the pipeline is the structure building module, which uses the information from the previous steps to construct a 3D model structure protein of the query sequence. This network will give you a single model, without any energy optimization step. Model building is based in a new concept of 3D structures generation, named IPA (Invariant Point Attention) and the use of a curated list of parametrised list of torsion angles to generate the side chains.

Like for most of the previous methods Alphafold would give your better results with proteins with related structures known and with a lot of homologs in Uniref databases. However, comparing to nothing, it will likely give you (limited) useful results for the so-called “dark genome”. I work with phages and bacterial mobile elements, and sequencing that is often frustrating as more than 50% of the proteins have no homologs in the database. So you have a bunch of proteins of unknown function… However, as we do know that structure is more conserved than sequence, we may use the structure to find out the function of our dark proteins. There are a few resources for this, I’d suggest you to try FoldSeek (Kempen et al., n.d.) and Dali (Holm 2022) servers. You can upload the PDB of your model and search for related structures in PDB and also in Alphafold database.
As mentioned above, Colabfold aims to make the process faster by using MMSeqs in the first block. Additionally, the number of recycling steps can also be adapted. Moreover, different Colabfold notebooks have been developed (and evolved) to allow some customization and other feature, like batch processing of multiple proteins avoiding recompilation and identification of protein-protein interactions (Mirdita et al. 2022).
Alphafold models can be evaluated by the mean pLDDT, a per-residue confidence metric. It is stored in the B-factor fields of the mmCIF and PDB files available for download (although unlike a B-factor, higher pLDDT is better). The model confidence can vary greatly along a chain so it is important to consult the confidence when interpreting structural features. Very often, the lower confidence fragments are not product of a poor prediction but an indicator of protein disorder (Wilson, Choy, and Karttunen 2022).
Alphafold also partnered with EMBL-EBI and Uniprot and generated a huge curated database of proteins from model organisms (Varadi et al. 2022), the Alphafold database. This is an amazing resource that may be also very helpful for you. Just consider that this database increased from 48% to 76% the fraction of human proteome with structural data, and also it also means great increases in the case of other model organisms, like, including microorganisms and plants (Porta-Pardo et al. 2022).

Let’s try Alphafold2.
Section under construction!
As mentioned above, the grand breakthrough of Alphafold would not have been the same without the Colabfold, a free open tool that made the state-of-the-art of AI-fueled protein prediction available to everyone.

The Colabfold repository on GitHub contains links to several Python “notebooks” developed on Google Colab, a platform to develop and share Python scripts on a Jupyter Notebook format. Notebooks are very important also for reproducibility in computer sciences, as they allow you to have the background and details and the actual code in a single document and also execute it. You can share those notebooks very easily and also update quickly as they are stored in your Google Drive.
Colabfold allow you to run notebooks of Alphafold and RoseTTAfold for specific applications, allowing even to run a bunch of proteins in batch. You can see a more detailed description in Mirdita et al. (2022). We are using the Alphafold2_mmseqs2 notebook, that allow you most of the common features. You need to allow Colab to use your Google account.

Then paste your sequence and chose a name. For more accurate models you can click “use_amber” option. It will run a short Molecular Dynamics protocol that ultimately optimize the modeling, but it will also take some more time, so better try at home.
As you can see, an this is a recent feature, you can also add your own template. That will safe time, but of course without any guarantee. If you have a template of a related protein, like an alternative splicing or a disease mutant, I’d advise you to try with and without the template. You may surprise.
At this point, you may execute the whole pipeline or may some more customization. MSA stage can be also optimized to reduce execution time, by reducing database or even by providing your own MSA. Very often you may want to fold a protein with different parameters, particularly in the Advanced Colabfold, which may very convenient to reuse an MSA from a previous run (although they recently updated servers for MMSeqs and made it really faster). If your proteins are in the same operon or by any other reason you think that they should have co-evolved, you prefer a “paired” alignment. But you can always do both.
Advanced settings are specially needed for protein-protein complexes. Also the number of recycling steps will improve your model, particularly for targets with no MSA info from used databases. Then you can just get your model (and companion info and plots) in your GDrive or download it.
What do you think is the ideal protein for alphafold2? Do you think homology modeling is dead?
Corollary: Has Levinthal’s paradox “folded”?
The development of Alphafold and the Alphafold structures Database in collaboration with EMBL-EBI has been the origin of a New Era. Scientific publications and journals worldwide published long articles about the meaning of this breakthrough in science and its applications in biotechnology and biomedicine1 and DeepMind claimed to have Solved a 50-years Grand Challenge in biochemistry. The coverage of the protein structure space has been greatly increased (Porta-Pardo et al. 2022).
However, some scientists have claimed that Alphafold2 and RoseTTAfold actually “cheat” a bit as it does not really solve the problem but generate a deep learning pipeline that “bypass” the problem (Pederson 2021). In agreement with that, it has been shown that machine learning methods actually do not reproduce the expected folding pathways while improving the structures during the recycling steps Outeiral, Nissley, and Deane (n.d.).
In conclusion, I do believe that Levinthal’s paradox has not been (yet) fully solved, but clearly almost (Al-Janabi 2022), and solving it will probably reduce the limitations of Alphafold2. However, CASP15 is currently being held and maybe I will have to change my mind later this year.
Useful links
Introductory article to Neural Networks at the IBM site: https://www.ibm.com/cloud/learn/neural-networks
ColabFold Tutorial presented presented by Sergey Ovchinnikov and Martin Steineggerat the Boston Protein Design and Modeling Club (6 ago 2021). [video] [slides].
Post about Alphafold2 in the Oxford Protein Informatics Group site: https://www.blopig.com/blog/2021/07/alphafold-2-is-here-whats-behind-the-structure-prediction-miracle/
A very good digest article about the Alphafold2 paper: https://moalquraishi.wordpress.com/2021/07/25/the-alphafold2-method-paper-a-fount-of-good-ideas/
Post on the Alphafold2 revolution meaning in biomedicine at the the UK Institute for Cancer Research website: https://www.icr.ac.uk/blogs/the-drug-discoverer/page-details/reflecting-on-deepmind-s-alphafold-artificial-intelligence-success-what-s-the-real-significance-for-protein-folding-research-and-drug-discovery
A post that explain how Alphafold2 and RoseTTAfold code became publically available: https://www.wired.com/story/without-code-for-deepminds-protein-ai-this-lab-wrote-its-own/
References
References
Footnotes
https://www.bbc.com/news/science-environment-57929095
https://www.forbes.com/sites/robtoews/2021/10/03/alphafold-is-the-most-important-achievement-in-ai-ever/
https://elpais.com/ciencia/2021-07-22/la-forma-de-los-ladrillos-basicos-de-la-vida-abre-una-nueva-era-en-la-ciencia.html↩︎