BIBMS-3: StrBio Exercise
1 Goal
The aim of this exercise1 is to explore and compare 3D protein structures using PDB files. You’ll learn how to:
Retrieve and visualize protein structures.
Perform structural alignments.
Analyze binding pockets.
Build a homology model from a new sequence.
💡 Note: Please post any questions in the Moodle Q&A forum so everyone benefits from the discussion.
2 Background
Viral diseases like COVID-19 and influenza can’t be treated with antibiotics. However, drugs such as oseltamivir (Tamiflu) inhibit viral proteins like influenza virus neuraminidase (NA), which targets sialic acid.
In the Protein Data Bank (PDB), you’ll find structures of neuraminidases from various influenza strains. For this exercise, we’ll focus on two NA variants:
2HU4 (oseltamivir-sensitive)
3CL0 (oseltamivir-resistant)
3 Procedure
1. Look for those structures and compare them.
- First, try to inspect them visually and find similarities and differences.
- Then, to quantify the similarity, you can then use the Structure Pairwise Alignment tool from the ANALYZE menu above and test different alignment algorithms (for more information on the algorithms check: https://www.rcsb.org/docs/tools/pairwise-structure-alignment) .
2. Now go to the full version of Mol* Viewer to analyze those proteins in more detail.
| You can download the structures directly from PDB database using the IDs. | 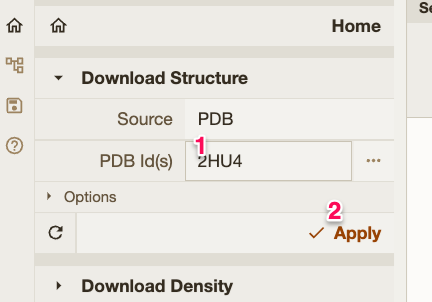 |
Zoom-in on the structures and see how each protein interacts with the inhibitor. Just move the mouse pointer over each residue and you will easily identify them by a label on the bottom-right corner. 💡 Tips:
|
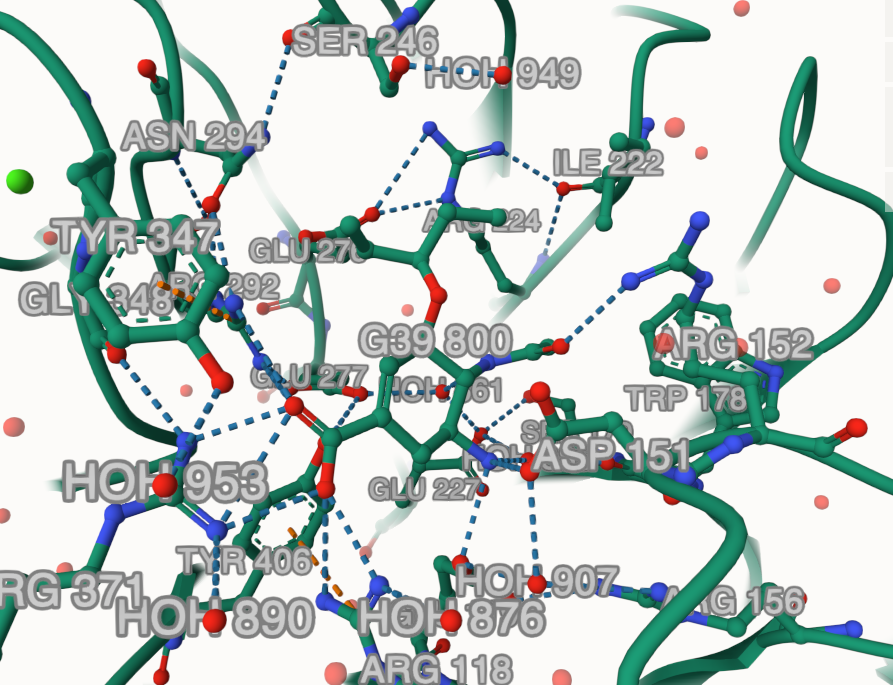 |
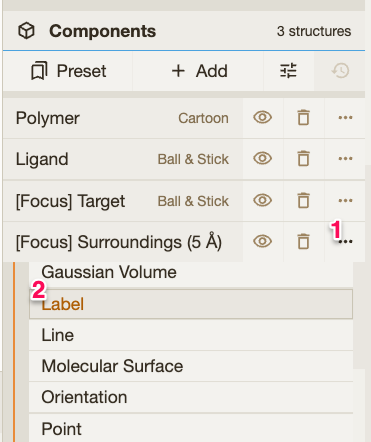
🪄 Trick: Click on the drug molecule and then use the right menu to change anything in the representation of the surrounding environment of the drug. For instance, you can add labels using Then click in the Do it for one monomer in each structure to see the differences in the binding pocket. |
 |
| You can also remove water molecules and extra ions that will not be useful for us at this point. | 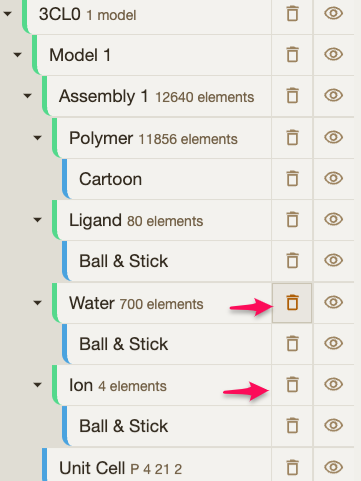 |
3. Now we want to compare them and check the details.
To align the structures: 1. Select the “Toggle” tool |
 |
| 2. Change the level for selecting to CHAIN |   |
3. Select one chain from each structure. 💡 Tip: Optionally, you can remove all the unwanted chains but one using the |
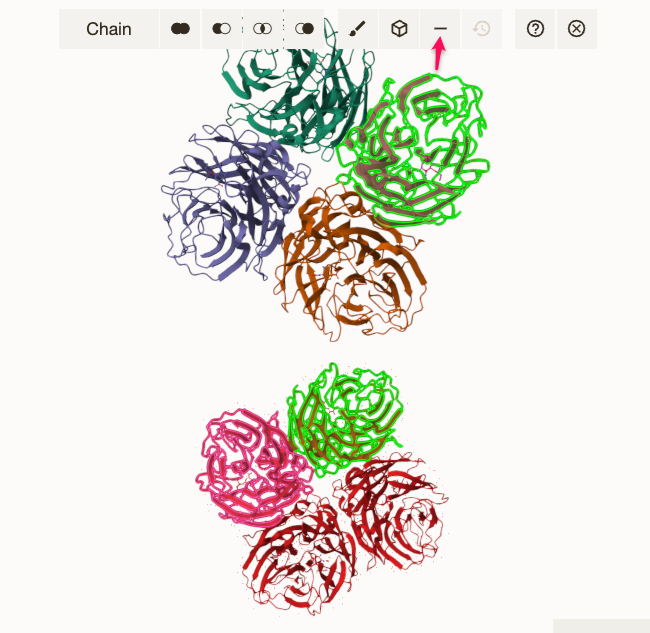 |
4. Align them with the The RMSD will be on the history bar (on the bottom). |
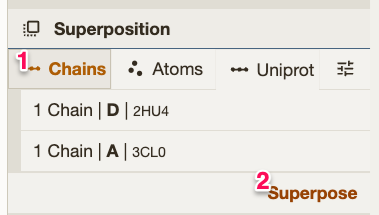 |
4. Compare the residues in binding pocket.
Focus especially on residues 274 and 276 in each structure.
5. We just found a new virus with the following neuraminidase sequence:
>New_neuraminidase
MNPNQKILCTTATAIVIGSIAVLIGIANLGLNIGLHLKPICNCSHSQPEATNASQTIINNYYNETNITQI
SNTNIQMEERASRGFNNLTKGLCTINSWHIYGKDNAVRIGENSDVLVTREPYVSCDPDECRFYALSQGTT
IRGKHSNGTIHDRSQYRALISWPLSSPPTVYNSRVECIGWSSTSCHDGKSRMSICISGPNNNASAVVWYN
RRPVAEINTWARNILRTQESECVCHNGSAVCPVVYTDGSATGPFDYRIYYFKEGKSALRWESLTGTAKWI
ERCSCYGERTGITCTCRDNWQGSNRPVIQIDPVAMTHTSQYICSPVLTDNPRPNDPNVGKCNDPYPGNNN
NGVKGFSYLDGVNTWLGRTISTASRSGYEMLKVPNALTDDRSKPIQGQTIVLNTDWSGYSGSFMDYWAEG
DCYRACFYVDLIRGRPKEDKVWWTSNSIVSMCSSTEFIGQWNWPDGAKIEYFLGSA- Try to obtain a structural model of that protein sequence.
- Check the target-template alignment and the model quality (Structure Assessment blue button).
You can use SWISS-MODEL: https://swissmodel.expasy.org/ All the top templates are very similar, thus you can pick the first one to construct your model. Once you construct your model, you can download it in PDB format. |
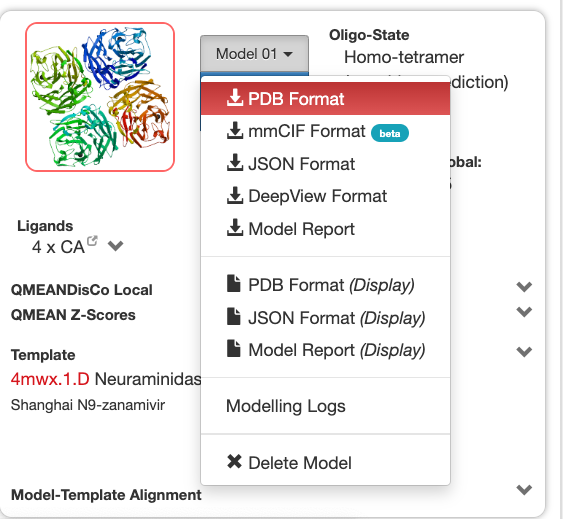 |
Import your model in Mol* using the Tip: Remember also to change back the selection to “Residue” and switch off the toggle option. |
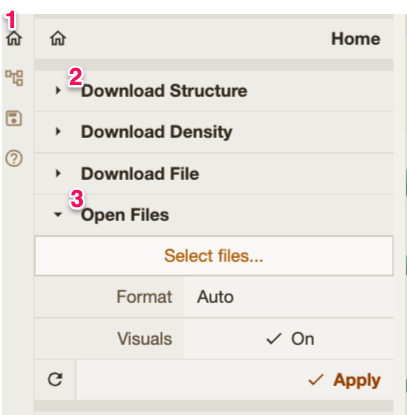 |
Help: Try to solve the whole exercise yourself, but if you have trouble comparing the three proteins, at this point you can view my work session containing the three structures: link You can import it using the OPEN option in the After opening the session you should rotate and select the drug molecules to show the binding pocket in the structures. Then you can select the residue you want to check. |
 |
Now, compare the position of the drug and interacting residues within the binding pocket in structures 2HU4 (sensitive) and 3CL0 (resistant) with the structure of the binding pocket in your model.
6. Go back to Moodle and answer the quiz entitled Structural Bioinformatics Assignment.
Footnotes
This exercise is inspired in the “Homology Modeling” exercise in the Course Bioinformatics & Molecular Biology by Department of Molecular Biosciences and Department of Informatics at the University of Oslo (UiO). However, this is a shorter and updated version adapted for BIBMS@UAM. Please let me know if you find any mistake or trouble.
 Distributed with CC-BY-NC-SA license.↩︎
Distributed with CC-BY-NC-SA license.↩︎